 |
||
Accueil > FORUM TERATEC > Programme > Atelier 4
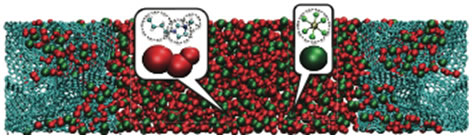
Modélisation par dynamique moléculaire de supercondensateurs à haute densité d'énergie Matthieu SALANNE Abstract : The recent demonstration that in supercapacitors ions from the electrolyte could enter sub-nanometer pores increasing greatly the capacitance opened the way for valuable improvements of the devices performances. Despite the recent experimental and fundamental studies on that subject, the molecular mechanism at the origin of this capacitance enhancement is still not quite clear. We report here molecular dynamics simulations including two key features: the use of realistic electrode structures comparable with carbide-derived carbons and the polarization of the electrode atoms by the electrolyte. This original design of an electrochemical cell allows us to recover capacitance values in quantitative agreement with experiment and to gain knowledge about the local structure and dynamics of the ionic liquid inside the pores. Then, from the comparison between planar (graphite) and porous electrodes, we propose a new mechanism explaining the capacitance enhancement in nanoporous carbons. We also set up some simulations where, starting from 0V, an electric potential is applied between the electrodes. It is then possible to follow the dynamical aspects of the charging of supercapacitors.
|

![]()
![]() La participation à cet atelier est gratuite sous réserve des places disponibles
La participation à cet atelier est gratuite sous réserve des places disponibles
![]() L'enregistrement en ligne est obligatoire pour y assister. Cliquer ICI pour vous inscrire.
L'enregistrement en ligne est obligatoire pour y assister. Cliquer ICI pour vous inscrire.
![]()